Chieh Lin (林潔)

PhD candidate of Machine Learning
Carnegie Mellon University
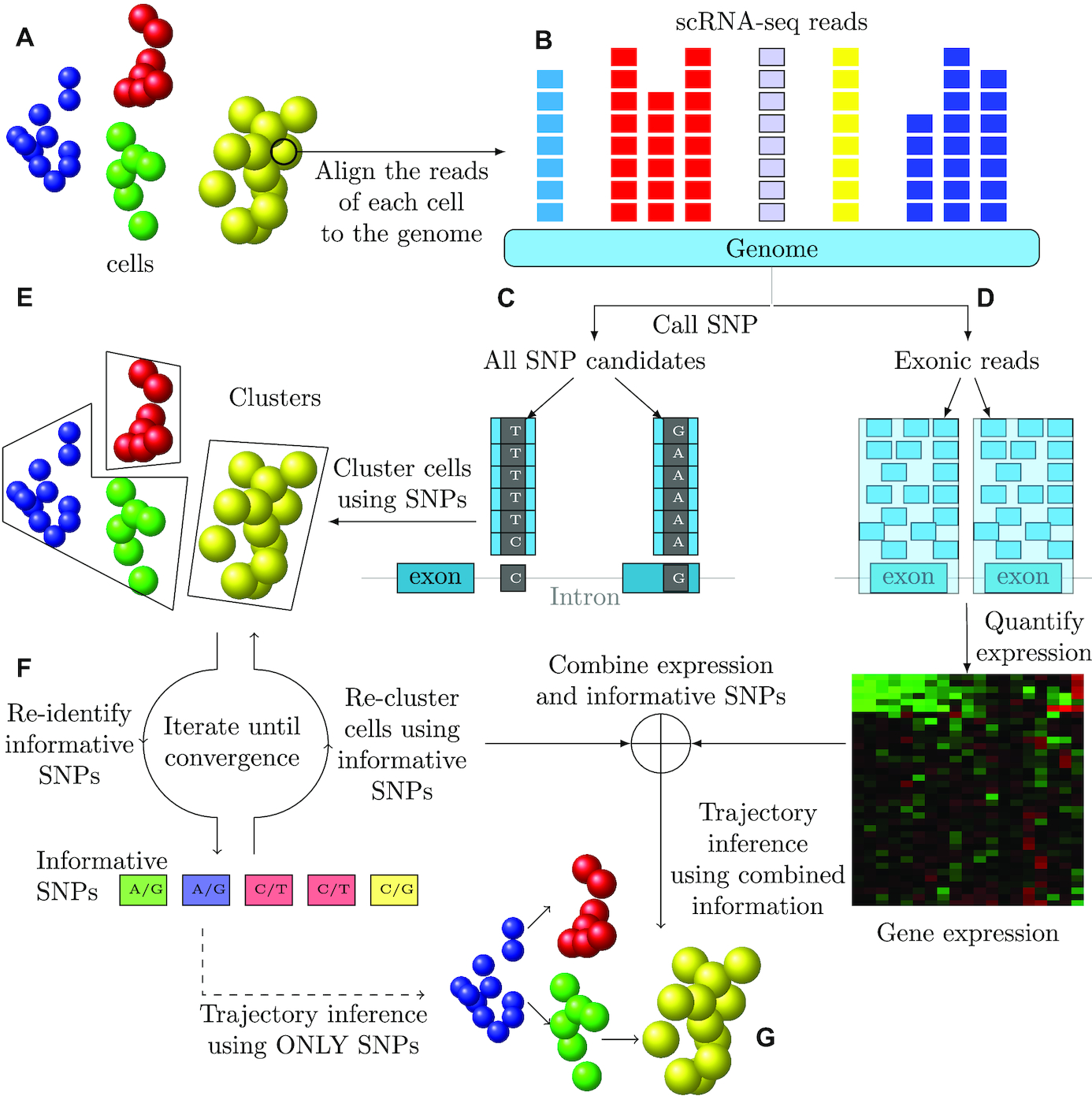
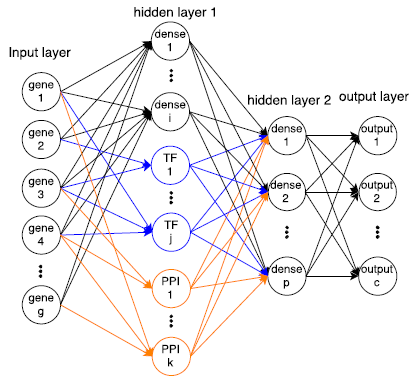
I am a fifth-year PhD student in Machine Learning at the Carnegie Mellon University, advised by Prof. Ziv Bar-Joseph , who leads Systems Biology Group. I plan to graduate before May 2020. My main area of interest is applying/designing Machine Learning models for solving real-world problems, including (but not limited to) improving the understanding of complex biological systems, drug discovery, healthcare problems, etc. My research with my advisor Ziv Bar-Joseph is mainly about designing new machine learning models to analyze time-series single-cell RNA sequencing data and find out potential biological insight in the models.
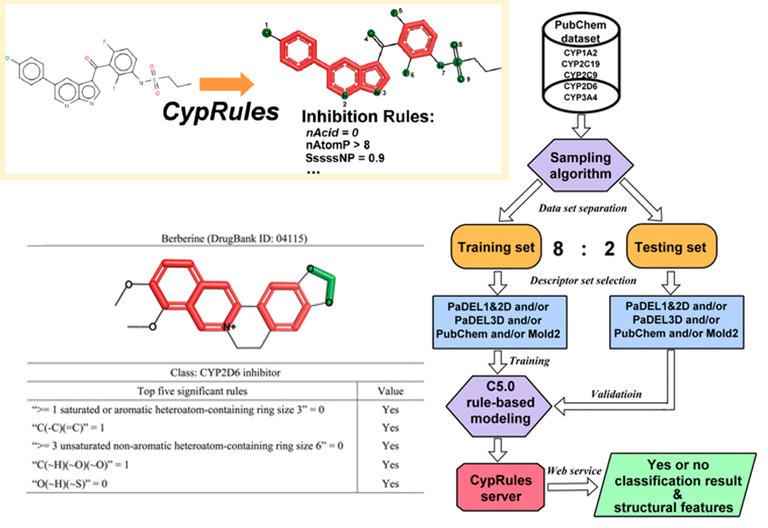
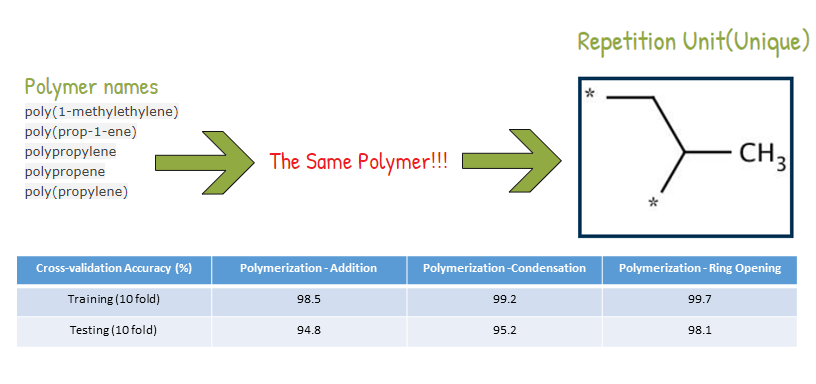
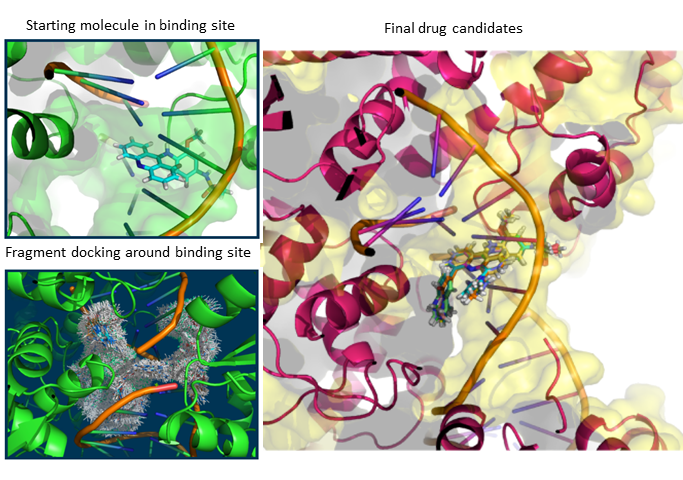
When I was a student in National Taiwan University, I joined Computational Molecular Design and Biomarker Detection in Metabolomics lab , lead by Prof. Yufeng Jane Tseng. My research projects with Prof. Tseng are mainly about using computational techniques for drug design and applying machine learning models in the Cheminformatics field.
Though my past research projects are either about biology or about chemistry, I am open to any other real-world problems.